-Clotting Factor Disorders-

-Three of the most common coagulation disorders are Factor VIII deficiency (Hemophilia A), Factor IX Deficiency (Hemophilia B) and von Wilebrands disease. These are X linked disorders
-The remaining coagulation disorders are autosomal recessive disorders
-The most common symptoms are excessive bleeding after procedures such as dental extractions and circumcision or mucosal bleeding such as epistaxis or menorrhagia.
-The performance of the PT and PTT is usually sufficient enough to identify these clotting disorders
-A prolonged PTT and normal PT=Factor XI deficiency
-A normal PTT and a prolonged PT is typical of Factor VII deficiency
-Prolongation of both PTT and PT suggests either a single deficiency of Factor X, V, prothrombin or fibrinogen or due to combination of these deficiencies
-Prothrombin Deficiency most commonly presents as excessive bleeding after invasive procedures such as umbilical cord, joint, muscles, or mucosal bleeding.
-Available treatments is Prothrombin Complex Concentrate (PCC) and Fresh Frozen plasma
-Factor V Deficiency is associated with minor to severe bleeding episodes. No specific concentrate is available. Treatment for severe bleeding is FFP.
-Treatment for combination of Factor V and VIII deficiency is FFP. There are no specific concentrate
-For Factor VII deficiency, it is hard to use FFP without causing volume overload. Factor VII has a short half life. Factor VII is ideal if available but not in US
-Recombinant Human Factor VIIa is available and approved in Europe for patients with Factor VII deficiency
-Recombinant Human Factor XIIIa is approved for treatment of Factor XIII deficiency in the United States
-Factor X Deficiency is treated the same way as prothrombin deficiency. Daily infusions of PCC or FFP is needed
-Prothrombin Complex Concentrates (PCC) is for treatment of factor IX deficiency, but also contains large amounts of II, VII and X and can be used to treat those deficiencies.
-Fresh Frozen Plasma is the mainstay of treatment for clotting disorders because it contains all the coagulation factors
-Factor XIII deficiency requires prophylaxis from the time of the diagnosis. The bleeding episodes are severe. FFP can also be given with the concentrate
-The Vitamin K Dependent Factors (Factor II, VII, IX, and X) deficiencies can be treated partially by administration of Vitamin K oral or parenteral. The severity of the bleeding episode depends on which route.
-Hypercoagulable States-

-The causes of thrombosis are divided into two groups inherited and acquired
-Virchow's Triad is a theory on the pathogenesis of thromboembolism: alterations of blood flow (stasis), vascular endothelial injury, and alterations in the constituents of the blood (inherited and acquired hypercoagulable states)
-Common inherited hypercoagulable states: Factor V Leiden, Prothrombin Gene Mutation, Protein S Deficiency, Protein C Deficiency, Antithrombin III Deficiency, and Dysfibrinogenemia
-Acquired risk factors include: more than 48 hours of immobility in the last month, hospitalization in the last 3 months, surgery in the last 3 months, trauma, IV drug use, pregnancy, oral contraception, tamoxifen, glucocorticoids, smoking with oral contraception, malignancy.
-Some patients with Antithrombin deficiency are resistant to heparin and antithrombin concentrate has been used safely
-Protein C Deficiency has been associated with warfarin induced skin necrosis but it is low, and is recommended to be used if no first degree relatives have a history of warfarin induced skin necrosis.
-Target treatment is with warfarin with a target INR between 2-3 for three to six months for initial DVT.
-Recurrence of DVT requires lifelong treatment with warfarin
-Pregnancy requires Lovenox because warfarin crosses the placental barrier. Heparin is also safe
-Thrombocytopenia-

-Thrombocytopenia is defined as a platelet count below the lower limit of 150,000.
-In general, surgical bleeding is a concern when the platelet count is less than 50,000.
-Severe spontaneous bleeding is a concern when the platelet count is less than 10,000
-Rarely, patients with thrombocytopenia are at risk for thrombosis rather than bleeding. Examples are below:
-Heparin Induced Thrombocytopenia there are antibodies to platelet factor 4 that cause thrombocytopenia, and platelet activation leading to life threatening arterial or venous thrombosis
-Antiphospholipid Syndrome is seen with patients with SLE, medications, and infection and cancer. Patients have arterial and venous thrombosis. Requires anticoagulation and treatment of the underlying condition
-Disseminated Intravascular Coagulation (DIC) are at risk for thrombosis and bleeding, usually venous. DIC patients are usually acutely ill with sepsis or malignancy
-TTP-HUS (Thrombotic Thrombocytopenia Purpura Hemolytic Uremic Syndrome) is associated with small vessel platelet rich thrombi. They can occur in any organ and can be life threatening. Treatment is plasma exchange and should be initiated promptly
-Paroxysmal Nocturnal Hemoglobinuria- is a rare condition caused by loss of glycosyl phosphatidylinositol from cell membranes. Can cause thrombosis of the intraabdominal veins and cerebral veins with hemolytic anemia and other cytopenias
-The major pathophysiologic mechanism of thrombocytopenia are: bone marrow problems, platelet destruction or consumption, and redistribution or splenomegaly
-Other causes of thrombocytopenia include: pregnancy, chronic liver disease or hypersplenism, immune thrombocytopenia, congenital platelet disorders, infections medications alcohol, malignancy, and nutrient deficiencies.
-Idiopathic Thrombocytopenia Purpura (ITP)-

-There are two criteria needed to make the diagnosis of ITP: the rest of the CBC including the blood smear is normal other than the thrombocytopenia and clinically apparent associated conditions (SLE, antiphospholipid antibody, and chronic lymphocytic leukemia) is not present
-The pathogenesis of ITP is related to a combination of increased platelet destruction and inhibition of megakaryocyte platelet production via specific IgG autoantibodies by the patients B cells often directed against the platelet membrane glycoproteins
-Splenomegaly is present because of sequestration
-Clinical manifestations include petechiae, purpura, easy bruising, epistaxis, gingival bleeding, menorrhagia. GI bleeding, gross hematuria, intracranial hemorrhage are rare
-Children with ITP typically present after infection
-Drugs that have been known to induce ITP include Alemtuzumab and purine analogs
-Initial treatments can include glucocorticoids and IVIG
-Splenectomy is definitive treatment for ITP
-Thrombotic Thrombocytopenia Purpura (TTP)-
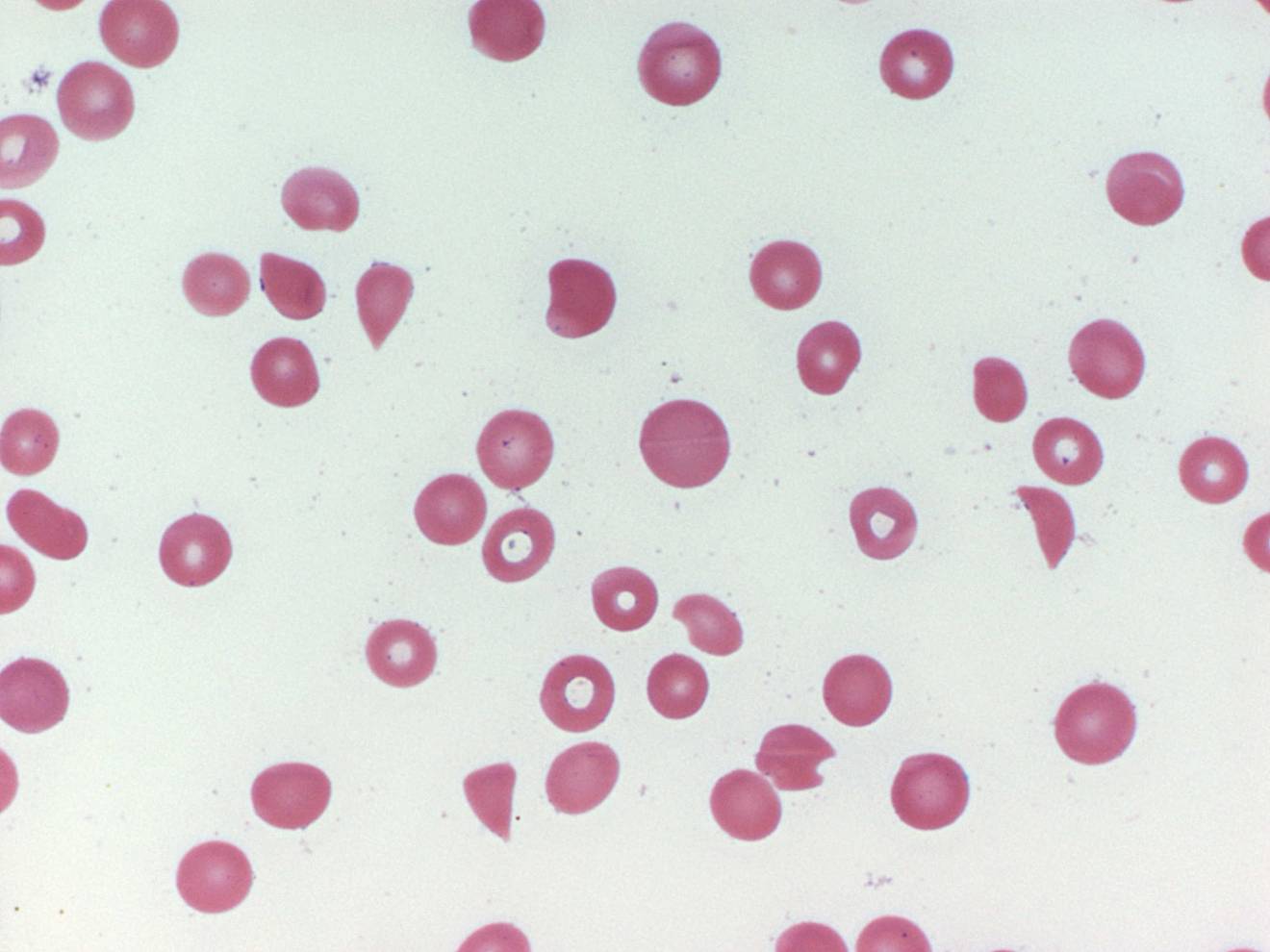
-Both TTP and Hemolytic Uremic Syndrome are both acute syndromes with abnormalities in multiple organ syndromes and evidence of microangiopathic hemolytic anemia and thrombocytopenia
-TTP and HUS and often discussed as separate syndromes the presenting features are almost the same in both adults
-In a few patients neurologic abnormalities are dominant and acute renal failure is minimal or not present
-When acute renal failure is dominant the disorder is usually considered HUS
-Clinical manifestations include: microangiopathic hemolytic anemia, thrombocytopenia often with purpura but not usually severe bleeding, renal function may be normal, but renal insufficiency may be present associated with anuria, neurologic abnormalities usually fluctuating are common, fever is rare
-The diagnosis of TTP and HUS is made clinically and only requires thrombocytopenia and microangiopathic hemolytic anemia without another clinically apparent etiology
-The diagnosis of TTP should prompt initiation of plasma exchange therapy which is life saving in this
No comments:
Post a Comment